Blog about new medical equipment
Blog about new medical equipment
")
")
")
")
")
")

MEDICAL EQUIPMENT: VIEWS AND FEATURES
A doctor’s main tool is his or her sharp mind. But, no matter how sharp your mind is, you will perform surgeries with a scalpel or a radio wave knife.
")
PRINCIPLE OF OPERATION OF ELECTROSURGICAL UNITS
Nowadays, surgery using high-frequency electrosurgical devices is one of the modern technologies
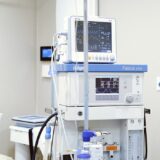
TYPES OF MODERN SURGICAL EQUIPMENT
Equipment for operating rooms and intensive care units can be classified according to their purpose: general surgical equipment – these are surgical instruments




 Best Online Casino GGBet: Official Club Evaluation Criteria
Best Online Casino GGBet: Official Club Evaluation Criteria
 Play at automatenspielex.com, the online casino where you can win real money!
Play at automatenspielex.com, the online casino where you can win real money!
 Step into the vibrant universe of online casinos at NettCasino.com; your gateway to unparalleled gaming and exhilarating moments!
Step into the vibrant universe of online casinos at NettCasino.com; your gateway to unparalleled gaming and exhilarating moments!
![]() Join Parimatch today and take advantage of our exclusive welcome bonus! Get started with a risk-free bet and start winning big.
Join Parimatch today and take advantage of our exclusive welcome bonus! Get started with a risk-free bet and start winning big.
![]() Experienced, caring and professional dentist in Surrey, BC. We serve adults and children, installing crowns and veneers.
Experienced, caring and professional dentist in Surrey, BC. We serve adults and children, installing crowns and veneers.
 Looking for non-stop action and endless thrills? Dive into the heart of the action at casino without Swedish license, and get ready to ride the waves of fortune at utansvensklicens.casino
Looking for non-stop action and endless thrills? Dive into the heart of the action at casino without Swedish license, and get ready to ride the waves of fortune at utansvensklicens.casino